Molecular Structure Inputs¶
htpolynet requires a molecular structure for every monomer (and any other small molecule) that appears in your system. It recognizes mol2 and pdb formats, and consumes them from ./lib/molecules/inputs/ relative to the directory in which you run htpolynet run or htpolynet parameterize. You can either let htpolynet generate these files for you from SMILES strings written directly into your configuration file (the recommended path, described next), or supply hand-prepared mol2/pdb files yourself (covered last). Either way, two very important considerations apply.
Before either path: valence conservation¶
htpolynet requires valence-conservation when polymerizing. When two atoms are identified as bonding partners (each typically on a separate molecule), each must own at least one sacrificial hydrogen that is deleted when the new bond forms, keeping the valence of each atom constant. We refer to this form of a monomer as its active form — and it is the active form that you describe to htpolynet, not the “actual” textbook form.
To illustrate, consider styrene. The “inactive” form is its actual structure:
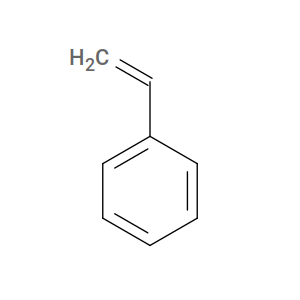
For htpolynet, however, styrene’s active form is ethylbenzene:
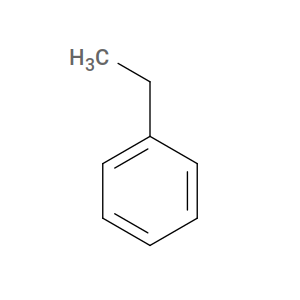
So the SMILES (or mol2) you give to htpolynet for styrene must describe ethylbenzene, with the two reactive carbons saturated and carrying the sacrificial hydrogens that will be removed when the inter-monomer bond forms.
In-config SMILES (recommended)¶
The simplest way to supply a monomer is to write its SMILES string directly into your configuration file inside the constituents block. htpolynet generates a 3-D structure, writes the mol2 to lib/molecules/inputs/<NAME>.mol2, and proceeds as if you had supplied the file by hand. Two paths are supported.
RDKit atom-mapping path (recommended; requires RDKit). Encode reactive atoms inline with SMILES atom-map labels (
[CH2:1]) and areactive_atomsmap keyed by those labels:constituents: STY: smiles: "c1ccccc1[CH2:1][CH3:2]" reactive_atoms: {1: C1, 2: C2}
This identifies the reactive atoms by chemical identity rather than by mol2 index, so the spec is robust to changes in atom ordering between toolchain versions. Install with
pip install 'htpolynet[smiles]'or use the container, which ships RDKit by default.Warning
SMILES bracket atoms (
[...]) take an explicit hydrogen count.[C:1]means zero implicit H — the carbon stays at its explicit valence. For an sp³ carbon you almost always want[CH:1](one implicit H) or[CH2:1]/[CH3:1]as appropriate. Mis-specified hydrogen counts typically show up as antechamber typing a saturated carbon asc2instead ofc3, propagating into a missing GAFF angle parameter intleap.OpenBabel index path (no Python extras). Provide
smilesand arename_atomsmap keyed by 1-based mol2 atom index:constituents: STY: smiles: "C1=CC=CC=C1CC" rename_atoms: {7: C1, 8: C2}
You need to know which
obabel-emitted indices to rename, which usually means runningobabelonce by hand to inspect the atom order.
In both cases htpolynet shells out to obabel to produce the final mol2 (RDKit itself has no mol2 writer), so OpenBabel must be on your PATH. See Installation and Prerequisites for setup details.
If a lib/molecules/inputs/<NAME>.mol2 is already present, it is left alone and SMILES regeneration is skipped — hand-edits survive a re-run. Delete the file to force regeneration.
Supplying mol2 or pdb files directly¶
If SMILES cannot cleanly capture your monomer (e.g., unusual stereochemistry, charged species, or coordinates from a published source), you can place a hand-prepared mol2 or pdb into lib/molecules/inputs/ directly. Two common ways to produce one:
Sketch and export. Any 2-D chemical sketcher that exports
mol2will work. For example, the ChemDoodle 2D sketcher: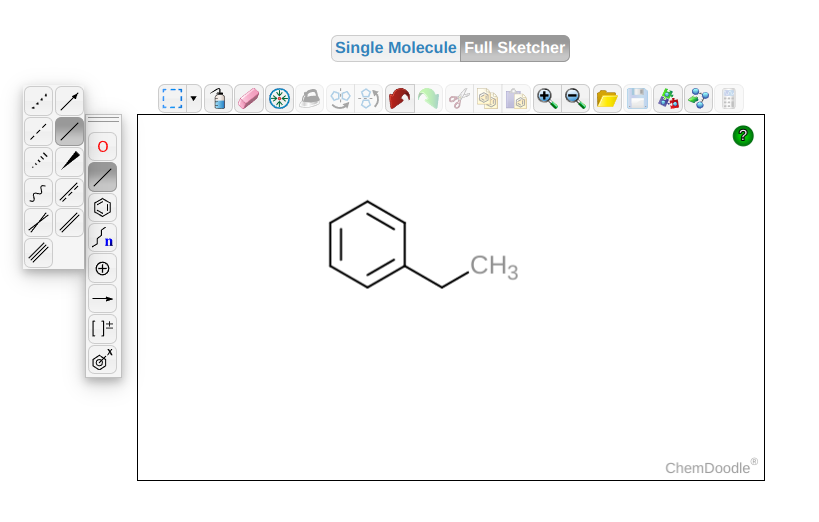
Fig. 2 Example of a ChemDoodle 2D-sketcher session for creating an input
mol2file for styrene (well, actually ethylbenzene).¶Standalone obabel. Write the SMILES on the command line and let
obabelproduce a 3-D structure:$ obabel -:"C1=CC=CC=C1CC" -ismi --gen-3d -h -omol2 -O STY.mol2
With either of these, you are responsible for editing atom names yourself (see below) before htpolynet can reference them.
Atom-naming¶
htpolynet expects every atom it must reference (i.e., every reactive atom) to have a unique name within its monomer. The names themselves don’t matter, only their uniqueness; non-reactive atoms can be left at their default names. The in-config SMILES paths handle this naming automatically via rename_atoms / reactive_atoms. For hand-prepared mol2/pdb files, you must edit the atom-name field yourself. Several atom-naming conventions are demonstrated in the tutorials.