Program Flow¶
flowchart TD
start(["<tt>htpolynet</tt> begins"]):::endpoint
pre["Make input<br/>molecular structures"]
setup["Set up reactions and<br/>oligomer templates"]
topo["Build system topology<br/><i>(e.g., init.top)</i>"]
coord["Build system coordinates<br/><i>(e.g., init.gro)</i>"]
md1["<b>MD:</b> Densification and<br/>precure equilibration"]:::md
cure[["<b>CURE</b>"]]:::cure
md2["<b>MD:</b> Postcure equilibration"]:::md
finish(["<tt>htpolynet</tt> ends"]):::endpoint
start --> pre --> setup --> topo --> coord --> md1 --> cure --> md2 --> finish
click cure "#cure-section" "Jump to CURE algorithm details"
classDef endpoint fill:#fff,stroke:#2a7a3f,color:#2a7a3f
classDef md color:#1f4e9c,stroke:#1f4e9c
classDef cure fill:#d4edda,stroke:#2a7a3f,color:#1e5128
Fig. 3 htpolynet run workflow.¶
A basic depiction of the the workflow initiated by htpolynet run is shown in the figure above. The first step is generation of the molecular structure data for any monomeric reactants, which htpolynet does using RDKit. Then, based on instructions in the configuration file, htpolynet proceeds with setting up all reactions and oligomer templates. Once these are generated, it then generates the full initial system topology in Gromacs format (that is, it generates a top file), and an initial set of coordinates (a gro file). Since the initial coordinates are built a low density (typically), htpolynet then performs MD to “densify” the system, followed by any pre-cure equilibration the user would like. Then the CURE algorithm takes over to generate intermolecular bonds and drive the polymerization. Once it finishes, htpolynet conducts any post-cure equilibration the user likes, before saving the final top and gro file. All of this work happens in the project subdirectory of the current directory in which run is invoked.
This workflow should make clear that the two required tasks of the user are:
The Connect-Update-Relax-Equilibrate (CURE) algorithm¶
flowchart TD
start(["<tt>CURE</tt> begins"]):::beginEnd
conv{"Conversion<br/>reached?"}
init["Initialize<br/>search radius"]
ident["Identify<br/>allowable bonds"]
found{"Any bonds<br/>found?"}
inc["Increment<br/>search radius"]
maxr{"Above max<br/>radius?"}
longer{"Any longer<br/>than threshold?"}
drag["Drag"]
topo1["Update topology"]
relax1["Relax"]
equil["Equilibrate"]
cap["Cap unreacted<br/>groups"]
topo2["Update topology"]
relax2["Relax"]
finish(["<tt>CURE</tt> ends"]):::endEnd
start --> conv
conv -- n --> init --> ident --> found
found -- n --> inc --> maxr
maxr -- n --> ident
maxr -- y --> cap
found -- y --> longer
longer -- y --> drag --> topo1
longer -- n --> topo1
topo1 --> relax1 --> equil --> conv
conv -- y --> cap
cap --> topo2 --> relax2 --> finish
classDef beginEnd fill:#d4edda,stroke:#2a7a3f,color:#1e5128
classDef endEnd fill:#fadbd8,stroke:#a93226,color:#7b241c
Fig. 4 Block flow diagram of the CURE algorithm used in htpolynet.¶
The algorithm used to create new bonds and polymerize a system is called the CURE algorithm, depicted above. This is just a slightly modified version of a standard search-radius-type algorithm, first used by Li and Strahan to study EPON/DETDA thermosets (Li and Strachan [LS10]). The CURE algorithm begins by executing a search for new bonds on a frozen system configuration. Bonds are downselected through a series of filters to arrive at a final set of bonds to form. If the distance between any pair of “bond-designate” atoms is greater than some threshold (the trigger_distance parameter in the drag subdirective of the CURE directive of a configuration file), a series of MD simulations that slowly bring all to-be-bound atom closer together is performed. Then the topology is updated, where htpolynet applies the charges, atom type, and bonded interaction templates from the oligomer template set to each bond. After the update, a series of relaxation MD simulations bring all bonds to their equilibrium lengths. Then a short NPT MD simulation equilibrates the overall density before initiating the next CURE iteration. CURE iterations continue until (a) a desired conversion is reached, or (b) no new allowable bonds are identified.
Identifying allowable bonds: Bondsearch filters¶
A key task of htpolynet is identifying potential bonds between reactive atoms. htpolynet organizes its “bondsearch” in any one CURE iteration along reactions. Each reaction specified in the configuration file usually defines one bond by the identies of each reactant and the name of each atom in each reactant. The bondsearch algorithm begins considering each reaction, and for each one, filtering the set of possible bonds using a series of rules, depicted below.
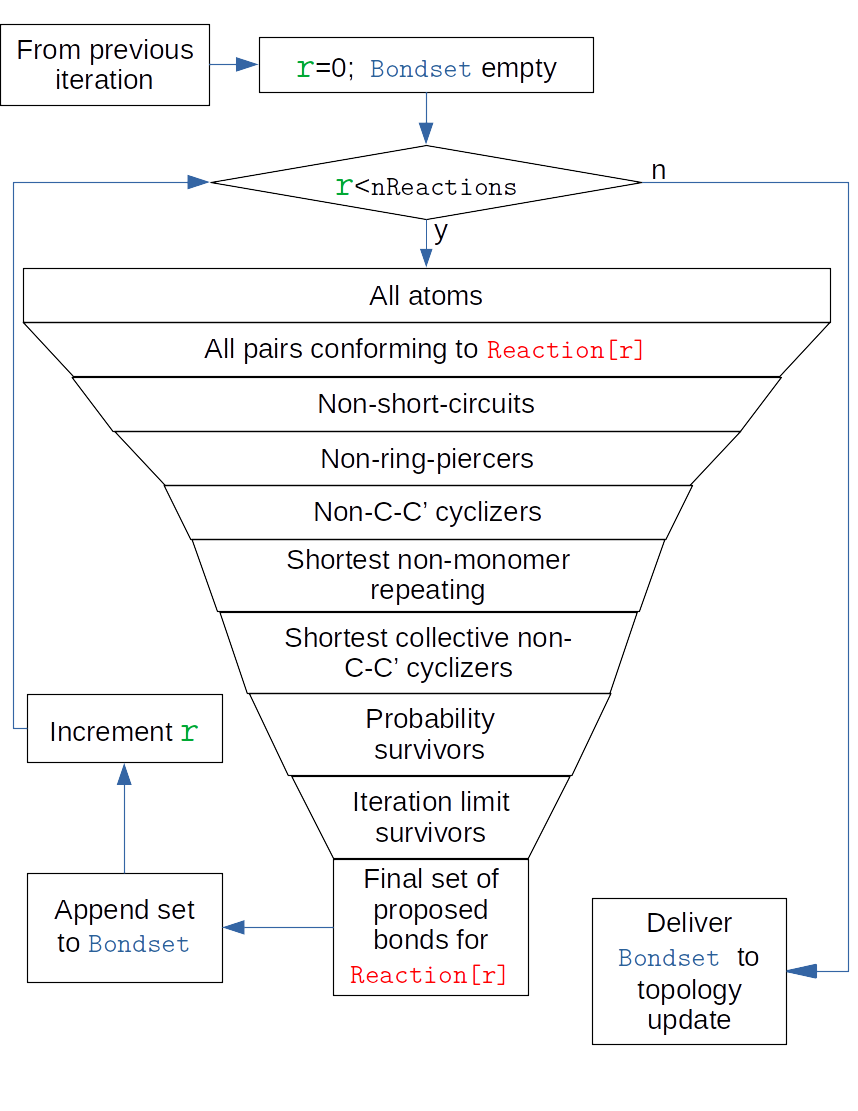
Fig. 5 Bondsearch filtering in one CURE iteration.¶
The outer loop in this figure corresponds to an iteration over reactions. An iteration consists of downselecting from all atoms to a set of pairs of atoms such that each member corresponds to one of the two atoms referred to in that reaction’s bond record. This selection is also limited to those atoms that still have sacrificial hydrogens. From this set, any potential bond that would result in a “short-circuit”, defined as two monomers that share more than one intermonomer bond, is excluded. Then any potential bond that “pierces” a ring is excluded. In the case that the polymerization chemistry involves opening of C-C double bonds, any potential bond that results in a “cycle” of C-C bonds of any length is excluded. This now smaller set of potential bonds are then sorted by length and the topmost potential bonds (shortest) that do not repeat any monomer index are retained. Then the entire set of potential bonds is considered at once to determine if any cycles of C-C bonds would form, and if so, the longst potential bond in any cycle is disallowed. Then, for each bond, a random number between 0 and 1 is drawn and compared to its assigned probability; if the random draw is greater than the probability, the bond is disallowed. Finally, if the current allowable conversion in the iteration is limited by a user directive, the longest bonds beyond this limit are also disallowed. This final set of bonds is forwarded to the topology update; if this set is of zero length, the topology update immediately hands off to the radius checker.